如何用云计算模式,实现生物信息分析 | 硬创公开课

雷锋网 (公众号:雷锋网) 按:当基因检测变得越来越普及,所得数据越来越多,对数据解读的需求也由此诞生,而这就是生物信息技术的作用。本文整理自华点云技术总监于伟文在雷锋网硬创公开课上的演讲,主题为如何用云计算模式实现生物信息分析。
于伟文,高级工程师,上海华点云生物科技有限公司技术总监。是北京航空航天大学国家示范学科——“移动云计算”专业的首批毕业生。拥有十余年 IT 领域从业经验,主要关注于临床、生物、金融等领域。作为课题负责人,曾主持过国家“十一五” 重大专项子课题;参与多项国家自然科学基金、国家重点实验室专项信息化建设项目。在Nature 子刊、SCI等权威刊物上发表学术论文10 余篇。
公开课视频如下:
以下为雷锋网整理的演讲主要文字内容。
华点云专注于高通量基因数据分析的公司,研发了一系列基因高通量生物分析工具,有基于NGS的数据传输、分析、报告、解读为一体的云端协同分析解决方案。这次的公开课则主要讲讲,如何用云计算模式实现生物信息分析。这次公开课分四部分:
一是生物信息学发展的历程,以及当前面临的挑战。获取数据的成本越来越低,数据量越来越大,现在的生物信息学面临的痛点,包括数据传输、计算及安全等方面。今天主要讲高性能计算的痛点。
二是分析NGS标准的生物信息分析的模式,中间数据处理的格式是怎么样的。你可能知道,科研单位主要是单样本分析,时间长且占资源。企业一般是多样本分析,痛点是NGS分析流程比较复杂,分析时间长,对集成带来压力。现在有两种计算模式:一种是弹性计算,基本能解决八成的问题;二是分布式计算。
三是云计算模式的特点与优势。
第四部分会以我们的乳腺癌检测产品为例,看如何在云端实现生物信息分析流程。

1953年,沃森和克里克提出了DNA 双螺旋结构,标志着生物科学的发展进入了分子生物学阶段,使遗传的研究深入到分子层次,“生命之谜”被打开,人们清楚地了解遗传信息的构成和传递的途径。这也开启了生物信息学的纪元,虽然当时还没这种叫法。

生物信息学之父是林华安,他提出了Bioinfomatics这种词。右图是人类基因组计算,自1990年正式实施,是生命科学的“登月计划”,也让生物信息学走向高潮。
人类基因组计划中有一个关键人物克雷格·文特尔,到1997年时整个计划只完成了3%,但时间和经费都用去了一半多。文特尔说有方法在3年内完成基因组测序,他以一己之力单挑6国科学家,采用“鸟枪法”以更快的速度完成人类。他本想为自己的方法申请专利,但在克林顿总统的要求下和其它6国科学进行了合作。
那什么叫测序?简单来说昌通过中间的测序仪,可以产生ACTG这样的数据,而生物信息学就是分析这些数据。
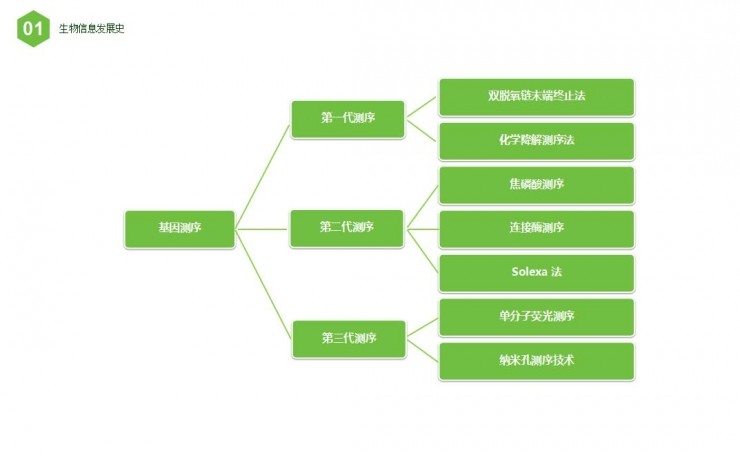
基因测序分为三代。
第一代测序技术的主要特点:优点是测序读长可达1000bp,准确性高达99.999%,从头测序,从头组装;而缺点是测序成本高,通量低,难以大规模的应用。
第二代测序的特点:大大降低了测序成本的同时,大幅提高了测序速度,维持了高准确性,但序列读长方面起第一代测序技术则要短很多。
第三代测序的特点:不需要PCR,避免了PCR错误的引入,读长可达3k,且速度快。
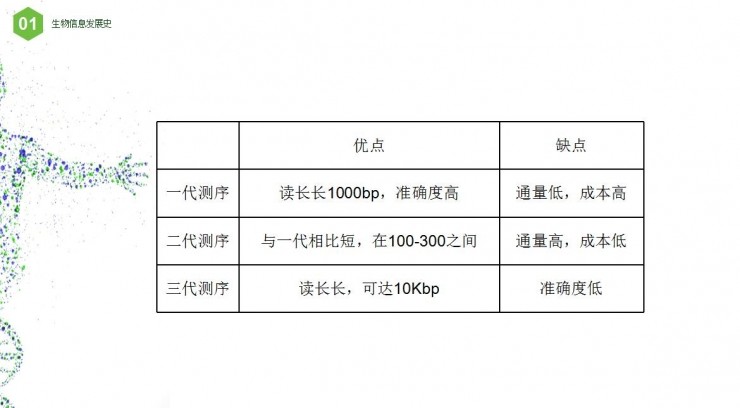
目前第二代测序是行业主流,后面的分析也是基于第二代。总结如上图。
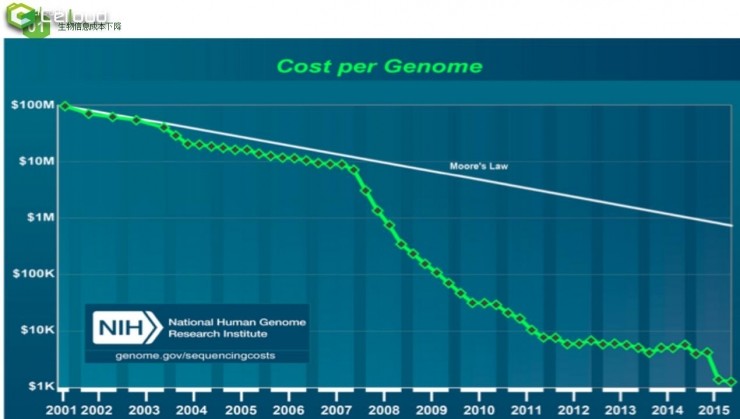
上图是测序成本的趋势。在2001年时测一个人的基因组花费近1亿美元,而2015年时只需要1000美元。
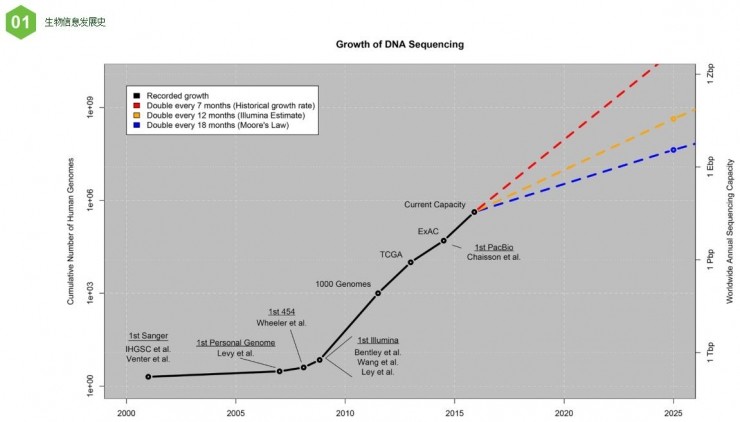
上图左侧是人基因组个数,右侧表示数据量,横坐标是时间。图中黑线是真实统计数据,蓝色是摩尔定律预测数据,红线是历史推测。可以看出,从2017年开始,每年的基因组数据会呈线性增长。

接下来看下数据量的大小。直接看图最后的数据,用最新的测序仪,在19-40个小时即会产生167GB到6TB数据,这还只是一台测序仪。
因此,当前考虑的不是基因测序本身的成本,而是数据传输、存储与分析的成本。目前面临的挑战主要包括(公开课主要涉及高性能计算方面):
数据解读
大规模的存储:大规模的存储: 混乱、缺乏行业标准
大量的计算资源:数据量大,计算复杂,时间长、成本高昂
高效的数据传输
安全稳定的运行环境
数据压缩,这也是行业难点
NGS计算的模式与挑战
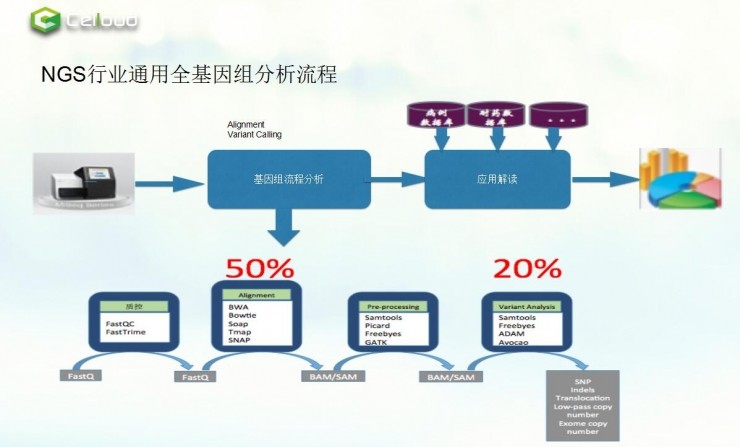
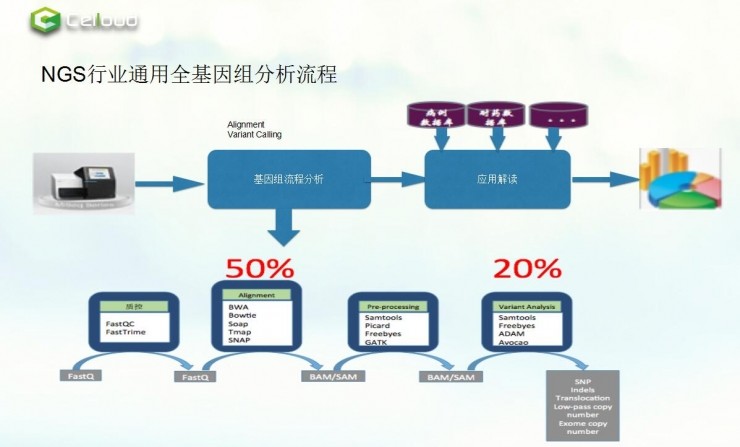
上图是NGS行业通用全基因组分析流程。测序后,通过标准化的基因组流程分析,然后与对应的数据库比对,就能形成标准的分析报告。
而标准的基因分析流程包括:质控,mapping,call SNP,注释。最重要的是mapping和call SNP,这是指它们对计算要求是非常高的。

AC和Trime是质控,当得到测序数据后,通过这两步把低质量数据去掉。mapping是指整个染色体数据拼成一条。最后就是去找突变位点在哪,找到定位后即可评估是什么样的病种,给出一个应对措施。
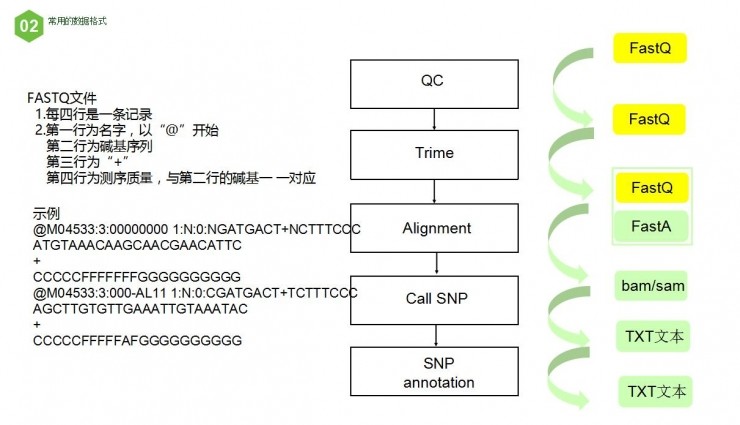
接下来看看每个阶段对应的数据格式是怎么样的。从QC到Trime都是FastQ格式,具体如上图所示。接下来会形成FastA格式。而通过mapping会再形成bam/sam格式。sam实质还是文本文件,只是对文件格式的定义;bam文件是对sam文件的二进制压缩,占用存储,后续处理速度快。通过call SNP则会生成文本。

接下来以微生物组学为例看NGS数据计算特征。如上图所示,微生物组学中,呈现单个样本数据量小、样本数量多,及单个样本数据量大,样本数量少等特征。同时看到,微生物组学与参考序列的比对过程中,计算量大。微生物组学全基因组数据的组装和聚类,对内存的消耗也很大。海量样本微生物组学数据的比较,也需要高IO。所以可以看到,整个NGS数据是计算密集型,内存密集型,IO交换密集型。
当前行业生物计算的业务场景是:
1.单样本数据量小,样本量很大:应该用弹性计算模式,能解决八成的需求。
2.单样本数据量大,样本量很小:采用分布式计算模式。
3.单样本数据量大,样本量较大:弹性+分布式计算模式。
云计算的特点与优势

整个云计算企业主要分为IaaS、PaaS、DaaS、SaaS。在云计算下整个医疗与健康厂商的划分如上,其中包括了各主要服务商。
云计算整个特点是:
成本低,以前完成一百个样本可能需要几万元,现在已经降到几元了。
弹性,即可以动态伸缩,满足动态的用户增长的需要。
以及高可靠性、通用性、虚拟化和超大规模等特点。
站在生物信息云端
第四部分最为重要,主要关于如何用云计算解决生物信息分析中存在的问题。

首先是“单样本数据量小,样本量大”的业务场景云端实现,这主要用弹性计算来实现。
上图是以乳腺癌检测应用为例,它基本代表整个NGS标准的分析流程。数据特点是单样本100M,有100个样本。
传统SGE集群计算模式单样本运行时间约15分钟,共约22小时,而我们的目标是15分钟全部完成。实现弹性计算,要要求业务需求选择云厂商,这里只需要从IaaS上选择即可。
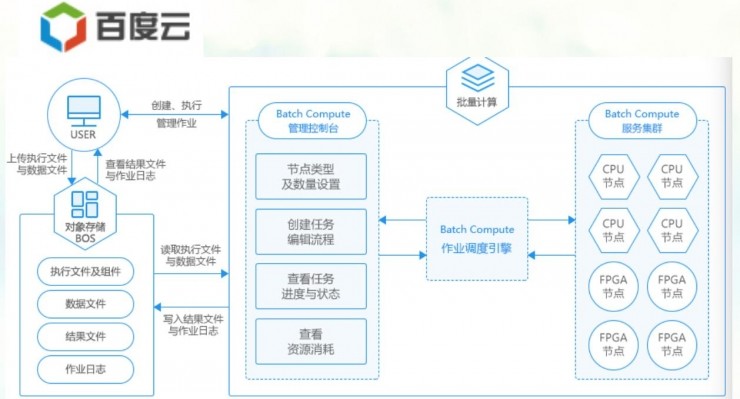
先看百度云,它提供批量计算。上图是百度技术架构图。第一步是先把数据传到对象存储BOS中,之后把作业投到Batch Compute计算集群里面。
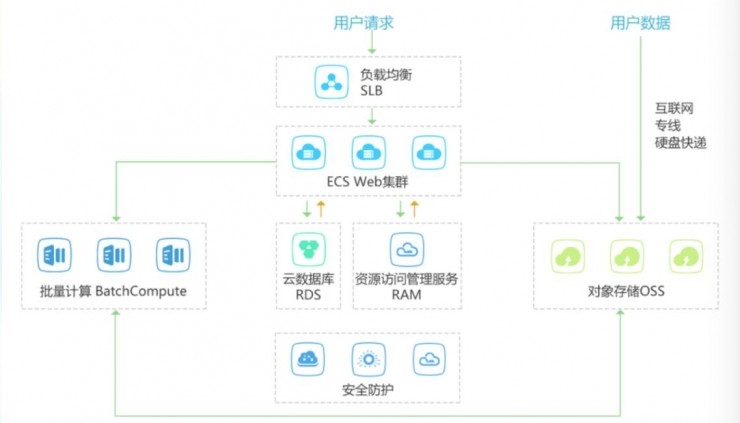
再看下阿里云,它也有Batch Compute计算模式,也适用于并行批处理作业的分布式计算。AWS由于网络问题,不太好用。
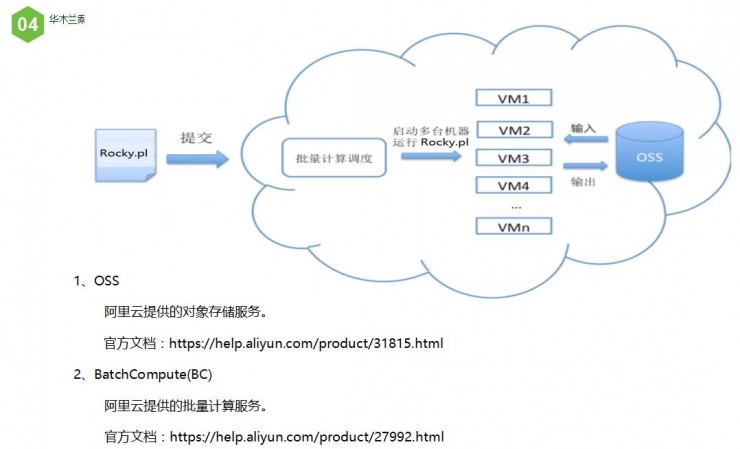
下面以阿里云为例,讲解如何去对Rocky.pl做弹性计算。做弹性计算,是让机房中的多台机器同时运行,把结果写到指定的OSS里。我们的任务写一个程序,快速让它在机房创建一排排机器(VM1-100),每个VM运行Rocky.pl,输入与输出都放到OSS上。这就是主要思路。
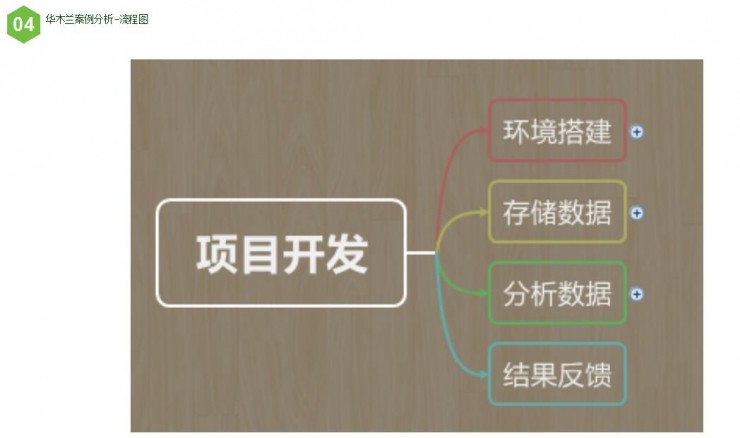
在写程序前,要做四件事,一是做环境搭建,二是把现有测试数据传到OSS上,三是正式写程序,四是查看测试结果。
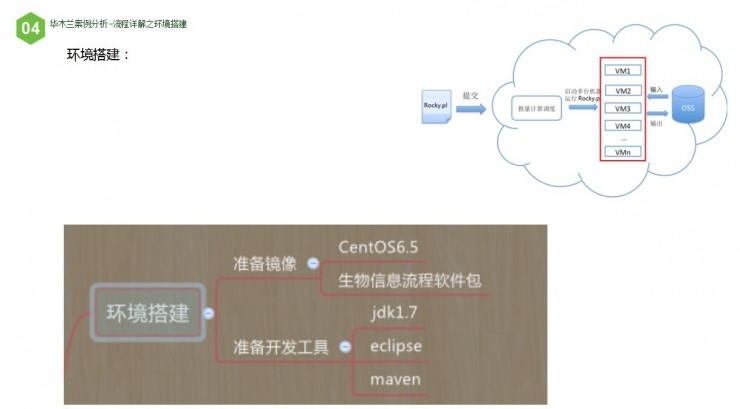
环境搭建第一步是创建虚拟机,让它运行rocky.pl的话,要先做镜像。例子中搭建的是CentOS6.5,当然这可根据需求自行决定,另外就是安装生物信息流程包。通过镜像可扩展100台机器。
第二是准备开发工具。这里以Java为例,需要的环境是jdk与eclipse。

准备工作完成后,要把数据传到OSS。刚才有三个文件,首先要将它们传到OSS。上传方式如上图所示,可以通过网页形式,或SDK的方式。
OSS中有几个概念。
一是bucket,即存储空间,用于存储对象(Object)的容器,所有的对象都必须隶属于某个存储空间。可以理解成windows下的C盘、D盘。
region是指地区,根据自己所在地域决定。
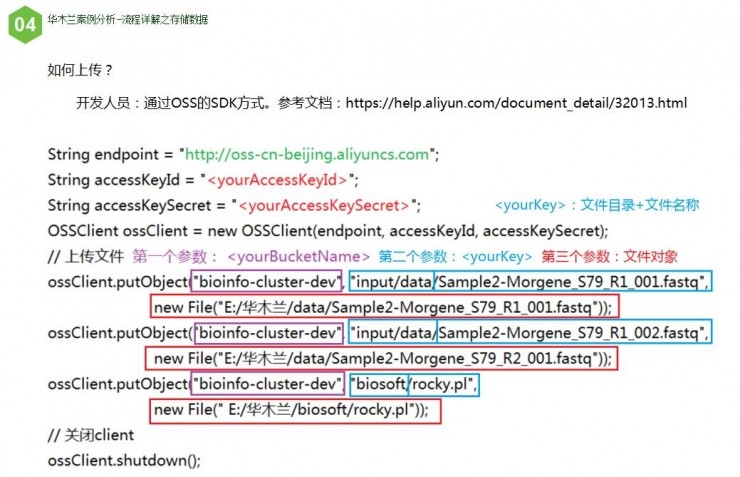
页面开发人员的SDK方式如上所示,需要输入账号密码,创建OSS客户端。具体细节不再详细展开。
接下来可通过SDK和控制台的方式查看是否上传成功。网络不稳定的情况下可选择断点续传。

下一步是写一个小程序,通过封装一步步实现弹性计算。做之前,要准备节点,让jar包中代码使用这个节点,这里会用到前面准备的镜像,并配置节点。然后是开发jar包,分别要构造job对象,构造DAG对象,构造task对象,以及OSS与节点的映射关系,最后封闭job的其他对象,并启动job。
这里有几个名词要解释。
job(作业)是BatchCompute的名词,每次分析操作都可以看成是一个作业。比如说我们要分析100个样本数据,那么当我们将100个样本当成一个整体,只需要执行一次,那么job就是1个。如果将这100个样本拆分成多个组,每组N个(N<100),那么就会有M个job(M=100/N)。例如每组5个样本,总共拆分成20个组,那么job个数就是20。
DAG是directed acycline praph的缩写,即有向无环图,通过它可以把业务流程切分成小的节点。
任务(task)也是BatchCompute的名词,是业务流程。我们可以将业务流程细分成N份,那么就可以有N个task。如果仅仅使用粗糙的业务流程,那么task设定成1即可。另外,每个job可以包含多个task,每个task单独用1台计算节点。

OSS与节点的映射关系,是将OSS上的数据挂载到节点上,以后此节点上的任何操作都是读取本地目录上的数据。这么做的好处在于减少了频繁和OSS进行交互,防止交互过于频繁,挂载连接断掉。而这种方式只需要最后关闭节点的时候,才和OSS交互。
原始DAG的缺点是,每台计算节点运算时间很短,但是等待资源时间很长。一般情况下,每个节点资源需要等待3-5分钟,而计算的时间可能仅仅几十秒。总的运行时间可能达到20分钟。

所以,一个想法是在一个task上运行所有步骤。要这么做,其中的步骤会如上图所示。

伪代码则如上所示,具体不再详细展开。程序非常简单,也有python和java等各个版本。
前面讲述的是一个job的运行流程。现在我们有100个样本,每个task运行2个样本,那么共需要50个task,每个task对应1个job,那么就需要50个job。测试阶段,我们可以使用for循环,执行50次。每个都运行一次job。伪代码如下:
for (int i = 1; i<=50; i++ ) {
//运行job
}
实际应用中,我们是通过一个调度系统,来判断当出现2个样本数据时,则创建一个job,并启动job,让这个job运行1个task,每个task里面执行2个样本数据。当有100个样本数据时,我们会启动50次job。伪代码如下:
//1.调度器发现有2个样本
//2.1构造job对象
//2.2构造task对象
//2.3将2个样本 set到task对象中
//3.创建job并启动

上图是真实测试数据,蓝色是开始时间,黄色是结束时间。可以看出,总耗时8分钟,费用是3元(阿里 云结果)。
用弹性计算方式,有两个问题。频繁对OSS读写,会造成OSS挂掉;另外,弹性资源有限制 ,比如阿里云限制是300台机器。
接下来是单样本数据量大,样本很小的业务场景,这又应该怎么去做呢?
再来看一下之前出现过的这一张图。如果单样本数据量大,相对它进行加速,不适用弹性计算,必须对NGS流程中的算法进行优化(如各流程中所示)。
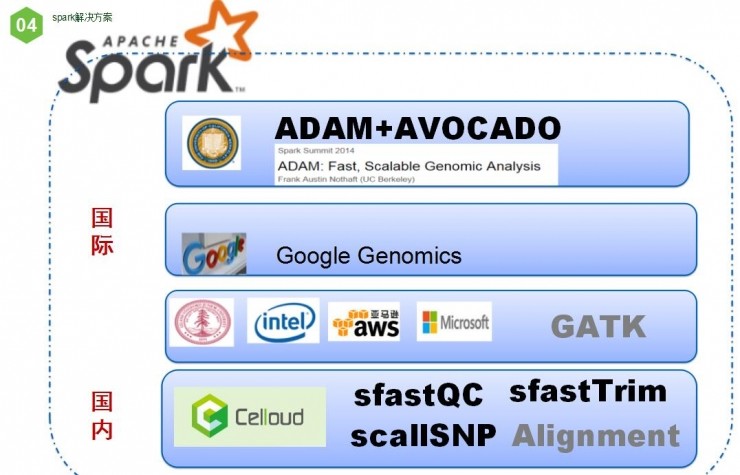
再来看看国际上,针对分布式计算的研究进展。Spark是伯克利大学的研究结果,而它还有一个ADAM的团队,它专门用于生物信息计算。Google有自己的Google Genomics,而英特尔、斯坦福和微软则是基于GATK进行加速,不过目前还没有成型。
国内目前没看到对生物信息用的算法进行加速,我们现在对NGS分析流程,基于分布式计算实现了加速。我们在fastQC、Trime、call SNP上都有进展。
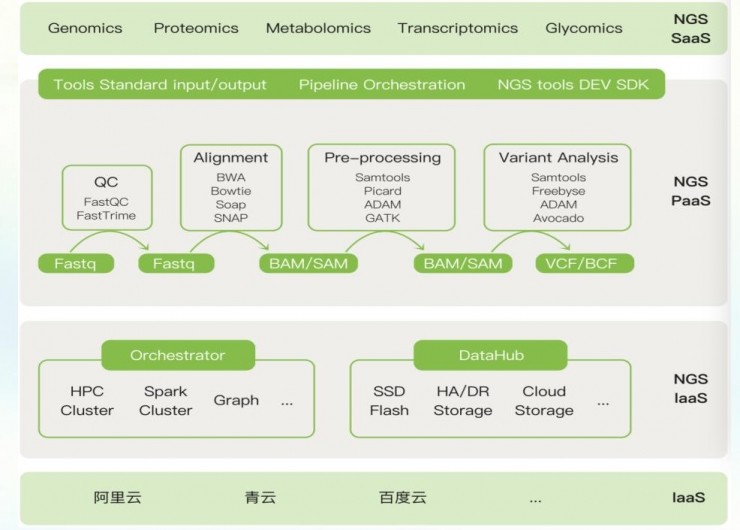
最后介绍一下弹性分布式计算的架构图应该是怎么样的。底层是公有云,在这些IaaS上,我们会搭建一个NGS的IaaS层,把适合于HPC、Spark、Graph的集群通过转换器封装起来成一个计算模式,同时把OSS、SSD等封装起来(DataHub),为上层的PaaS服务。
雷锋网原创文章,未经授权禁止转载。详情见 转载须知 。

正文到此结束
- 本文标签: 注释 百度 文章 IO 云 https 安全 数据 时间 总结 python 高通 虚拟化 ip centos UI 需求 开发 生命 Centos6 质量 集群 基金 代码 快的 http 人物 eclipse 产品 金融 IaaS 阿里云 ask 调度器 windows 压力 Region HTML 统计 Job 测试 数据库 配置 Google App src 1000美元 分布式 java 科技 安装 微软 空间 企业 2015 id 目录 map PaaS
- 版权声明: 本文为互联网转载文章,出处已在文章中说明(部分除外)。如果侵权,请联系本站长删除,谢谢。
- 本文海报: 生成海报一 生成海报二
热门推荐









相关文章






Loading...
![[HBLOG]公众号](https://www.liuhaihua.cn/img/qrcode_gzh.jpg)

