有参考基因组的RNA-seq详细分析流程
1. 简介
测序技术的普及使得RNA-seq进入寻常百姓家,单纯的qRT数据通量不再满足实验数据的需求,而RNA-seq的分析无非就是有参和无参两种方式;
本文主要就有参转录组的分析做简单介绍;
此外,有参转录组数据分析流程千千万,本文仅是其中一种,详细运行参数请多 -help ;

2. 环境准备
- 质量检验
- FastQC
- MultiQC (可选)
- reads 过滤与修剪
- Trimmomatic
- 序列比对
- hisat2
- 排序及格式转换
- samtools
- 序列组装
- StringTie
- 差异表达分析
- Ballgown
- DESeq2
- edgeR
3. 数据准备
- 目标物种基因组数据【基因组fa (genome.fa)和gff注释文件 (genome.gff3)】
- 测序 reads (实验室生成或NCBI下载)
4. 测序reads分析过程
4.1 SRA 转 fq (可选)
参考 Using the SRA Toolkit to convert .sra files into other formats ,根据个人喜好选用相应工具将NCBI的SRA数据库下载SRA数据转化为fq格式;
4.2 质控
FastQC 察看数据质量,Trimmomatic 来进行接头,低质量测序reads的过滤与修剪;
ls *gz |xargs -I [] echo 'nohup fastqc [] &'>fastqc.sh ./fastqc.sh multiqc ./
java -jar trimmomatic-0.30.jar PE / -threads 20 -phred33 reads1.fastq reads2.fastq / reads1.clean.fastq reads1.unpaired.fastq reads2.clean.fastq reads2.unpaired.fastq / ILLUMINACLIP:/Trimmomatic-0.30/adapters/TruSeq3-PE.fa:2:30:10 / LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50
4.3 序列比对
hisat2-build genome.fa genome hisat2 -x genome -1 read1.fq.gz -2 read2.fq.gz -S Sample.sam -p 8
4.4 排序及格式转换
samtools view -bS Sample.sam | samtools sort -@ 8 - Sample.sorted
4.5 序列组装
用StringTie对每个样本进行转录本组装
# Transcriptome assembly
stringtie -p 8 -G genes.gtf -o Sample.gtf –l Sample.sorted.bam
# 获取所有*.gtf 文件名的列表, 并且每个文件名占据一行
ls | /grep "Sample" | sort -V | uniq | awk 'BEGIN{OFS="/"} {print $1,$1".gtf"}' > Sample_gtf.txt
# Merges transcripts into a non-redundant set of transcripts
stringtie --merge -p 8 -G genes.gtf -o merged.gtf Sample_gtf.txt
# Expression level estimation
stringtie –e –B -p 8 -G merged.gtf -o Sample.gtf Sample.sorted.bam
4.6 count data 提取
准备上述gtf结果文件sample文件 (sample_lst.txt),格式如下:
Sample1 <PATH_TO_Sample1.gtf> Sample2 <PATH_TO_Sample2.gtf> Sample3 <PATH_TO_Sample3.gtf> Sample4 <PATH_TO_Sample4.gtf>
提取各样品count data
prepDE.py sample_lst.txt
5. 差异表达分析
差异分析可参考 搭PacBio全长转录组便车的无重复样本RNA-seq分析 ;
主要就是准备 表型文件和上述的基因或转录本count 文件 ;
表型数据格式如下 (phenodata.csv):
SAMPLE group Sample1 leaf Sample2 leaf Sample3 root Sample4 root
5.1 DESeq2
library("DESeq2")
countData <- as.matrix(read.csv("gene_count_matrix.csv", row.names="gene_id"))
colData <- read.csv(phenodata.csv, sep="/t", row.names=1)
all(rownames(colData) %in% colnames(countData))
countData <- countData[, rownames(colData)]
all(rownames(colData) == colnames(countData))
dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = ~ CHOOSE_FEATURE)
dds <- DESeq(dds)
res <- results(dds)
(resOrdered <- res[order(res$padj), ])
5.2 edgeR
edgeR 和上述 DESeq2相似,具体请参考其BiocManager 说明;
5.3 Ballgown
上述StringTie结果可直接用Ballgown读取来进行差异分析;
library(ballgown)
pheno_data <- read.csv("phenodata.csv")
bg <- ballgown(dataDir = "ballgown",
samplePattern = "sample",
pData = pheno_data)
samplesNames(bg)
bgfilt <-subset(bg,'rowVars(texpr(bg))>1',genomesubset=TRUE)(过滤掉表达差异较小的基因)
diff_genes <- stattest(bgfilt,feature='gene',covariate=【自变量】,adjustvars=【无关变量】,meas='FPKM')
diff_genes <- arrange(diff_genes,pval)
write.csv(diff_genes,'diff_genes.csv',row.names=FALSE)
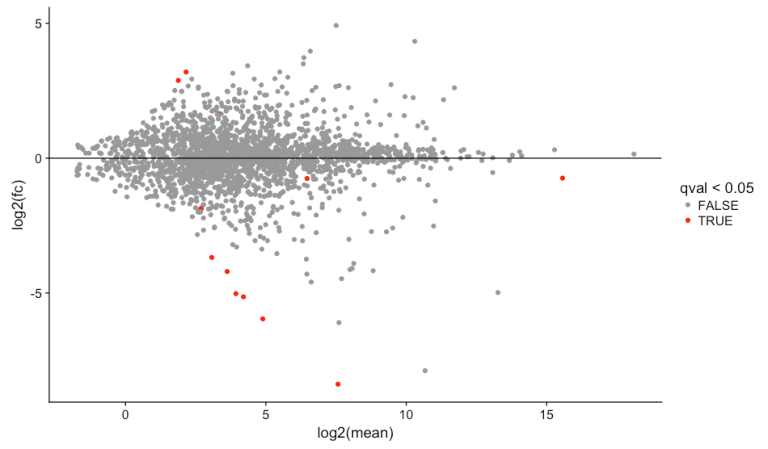
# MA plot
library(ggplot2)
library(cowplot)
results_transcripts$mean <- rowMeans(texpr(bg_chrX_filt))
ggplot(results_transcripts, aes(log2(mean), log2(fc), colour = qval<0.05)) +
scale_color_manual(values=c("#999999", "#FF0000")) +
geom_point() +
geom_hline(yintercept=0)
6. 扩增阅读
正文到此结束
热门推荐








相关文章

近期评论
-
谢谢
-
https://www.newcmy.com/register?aff=HBVX建议您试试草莓云机场,可以流畅观看youtube和tiktok,上reddit/x也没有问题,还有各种ai优化节点。
-
-
-
想购买您这个站,我的联系方式QQ741756694微信同步 能卖联系
-
-
-
-
-
https://www.liuhaihua.cn/archives/40657.html 这篇博客中的图片打不开了
Loading...
![[HBLOG]公众号](https://www.liuhaihua.cn/img/qrcode_gzh.jpg)

